Sequencing Errors
No sequencing technology is perfect, so all have some errors in reads. Next-Generation sequencing platforms differ in the rate, types, and randomness of their errors.
Truly random errors are the easiest to deal with. Given enough coverage, and a low enough error rate, these errors will disappear from a consensus sequence as the correctly called reads overwhelm the erroneous bases. PacBio has a high error rate (~10%) but claim that their errors are random.
Systematic errors are more of a problem. They’re likely to show up in the consensus sequence because they’re inherent to the technology, so if one read has a particular error, other reads are likely to have that same error. A well-known example of a systematic error is the homopolymer runs on 454 and Ion Torrent platforms; when more than 7 or so of the same nucleotide appear consecutively, the likelihood of a correct base call drops significantly.
Ion Torrent’s Strand-Specific Errors
In addition to the homopolymer issue, Ion Torrent has a second type of systematic error that isn’t as well-documented. I’ll call this the Strand-Specific Insertion/Deletion Error (S-SIDE).
The prototypical version of this error involves an area where the correct sequence should have a run of 3 Cs. On one strand, nearly all the reads will call this spot correctly, giving 3 Cs. On the other strand, however, almost all the reads will have only 2 Cs. (Click any image for a larger version.)
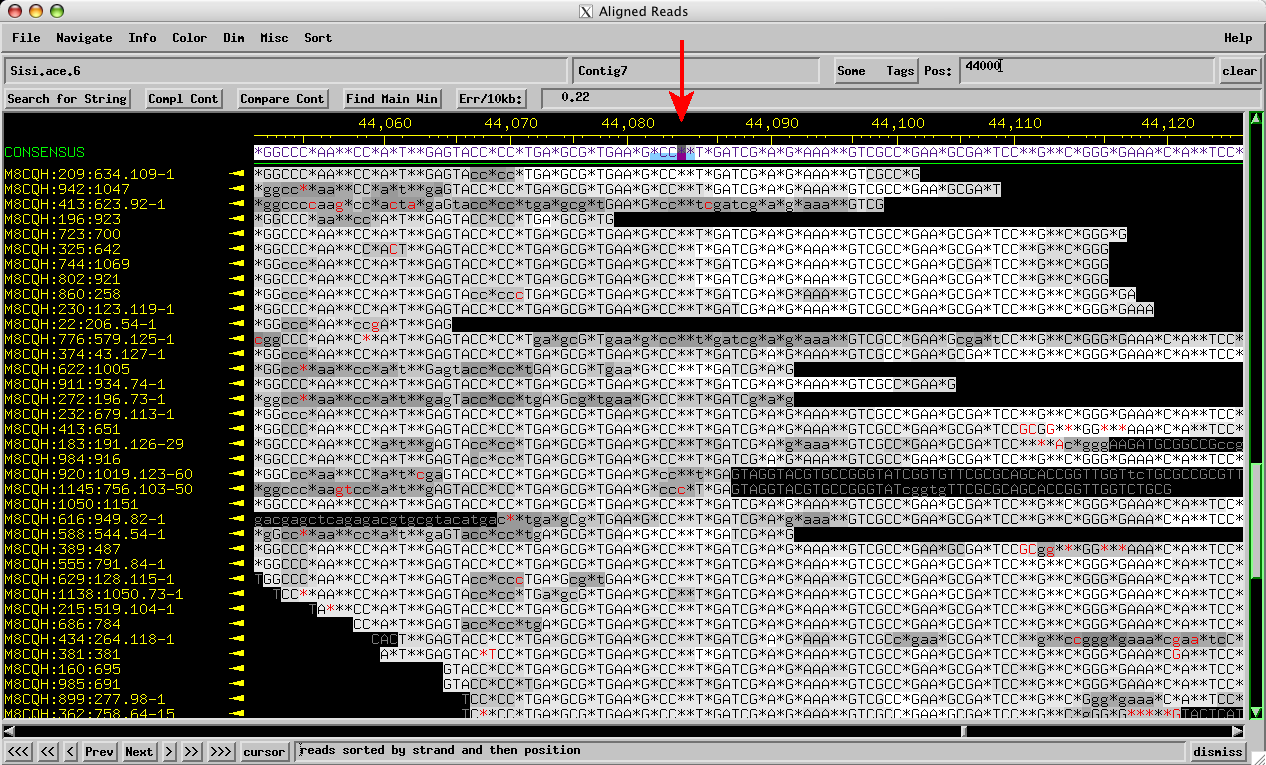 Figure 1: Sisi consed assembly reverse reads: Only 2 Cs called
Figure 1: Sisi consed assembly reverse reads: Only 2 Cs called
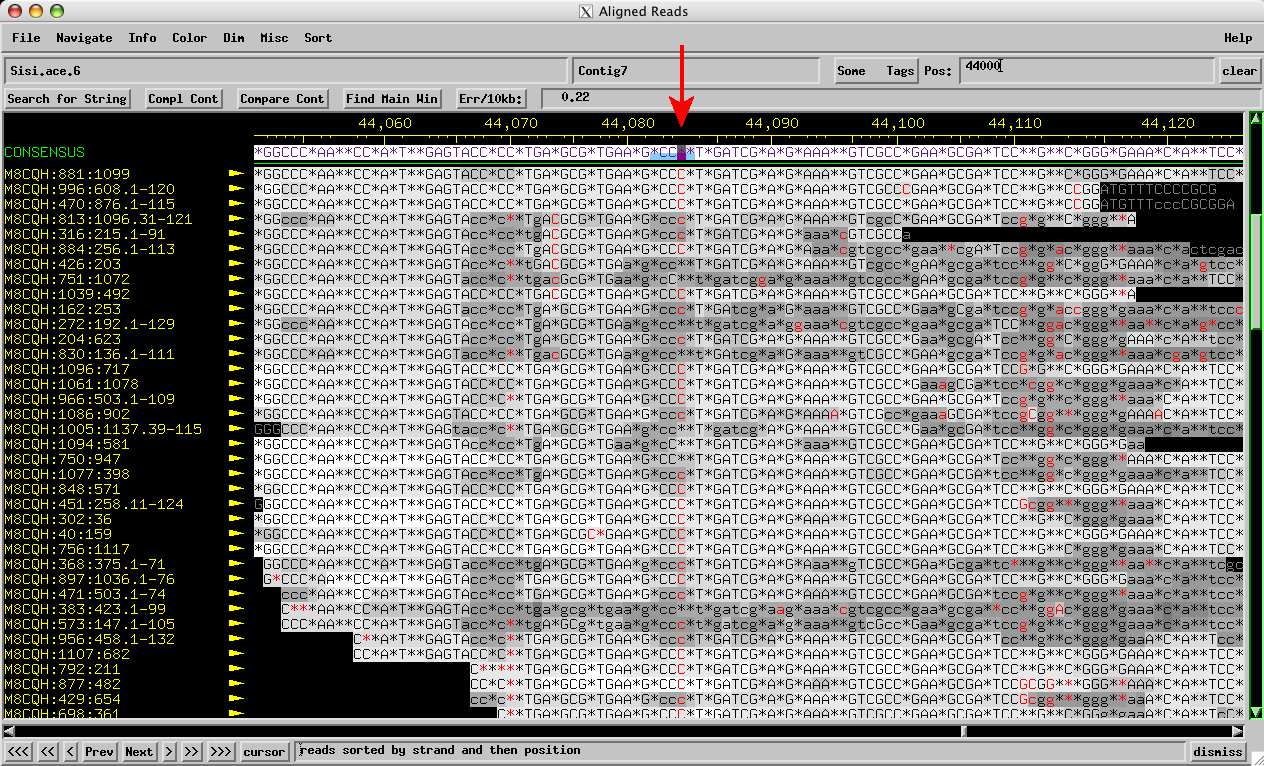 Figure 2: Sisi consed assembly forward reads: Three Cs called
Figure 2: Sisi consed assembly forward reads: Three Cs called
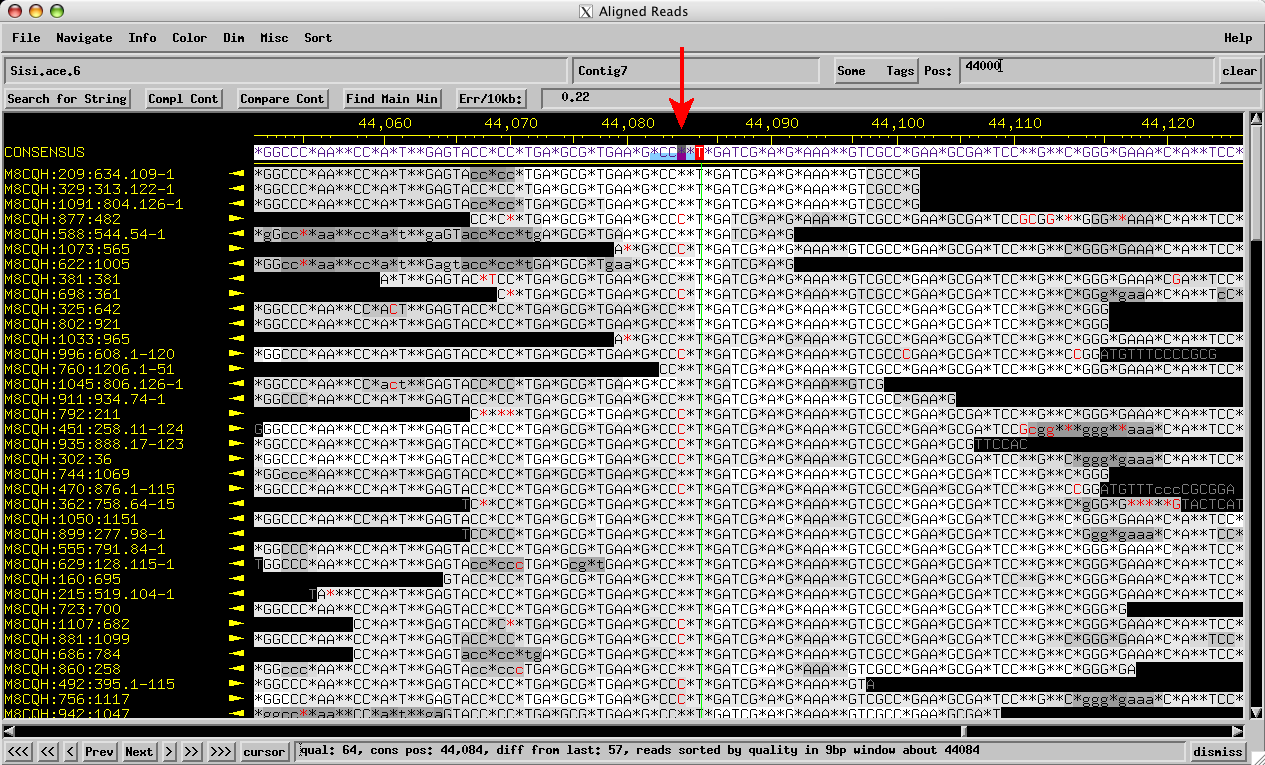 Figure 3: Sisi consed assembly mixed reads: ~Even numbers of reads with 2 Cs and 3 Cs
Figure 3: Sisi consed assembly mixed reads: ~Even numbers of reads with 2 Cs and 3 Cs
This error happens most often with Gs or Cs, but can also happen with As or Ts. It also is most common with runs of 3 nucleotides that are erroneously called as 2 on the opposite strand, but can also show up as:
- 4 called as 3 (GGGG vs GGG*)
- 2 called as 1 (GG vs G*)
- 3 called as 1 (GGG vs G**)
- 2 called as 0 (GG vs **)
- 1 called as 0 (G vs *)
 Figure 4: Milly consed assembly forward reads: 3 Gs called
Figure 4: Milly consed assembly forward reads: 3 Gs called
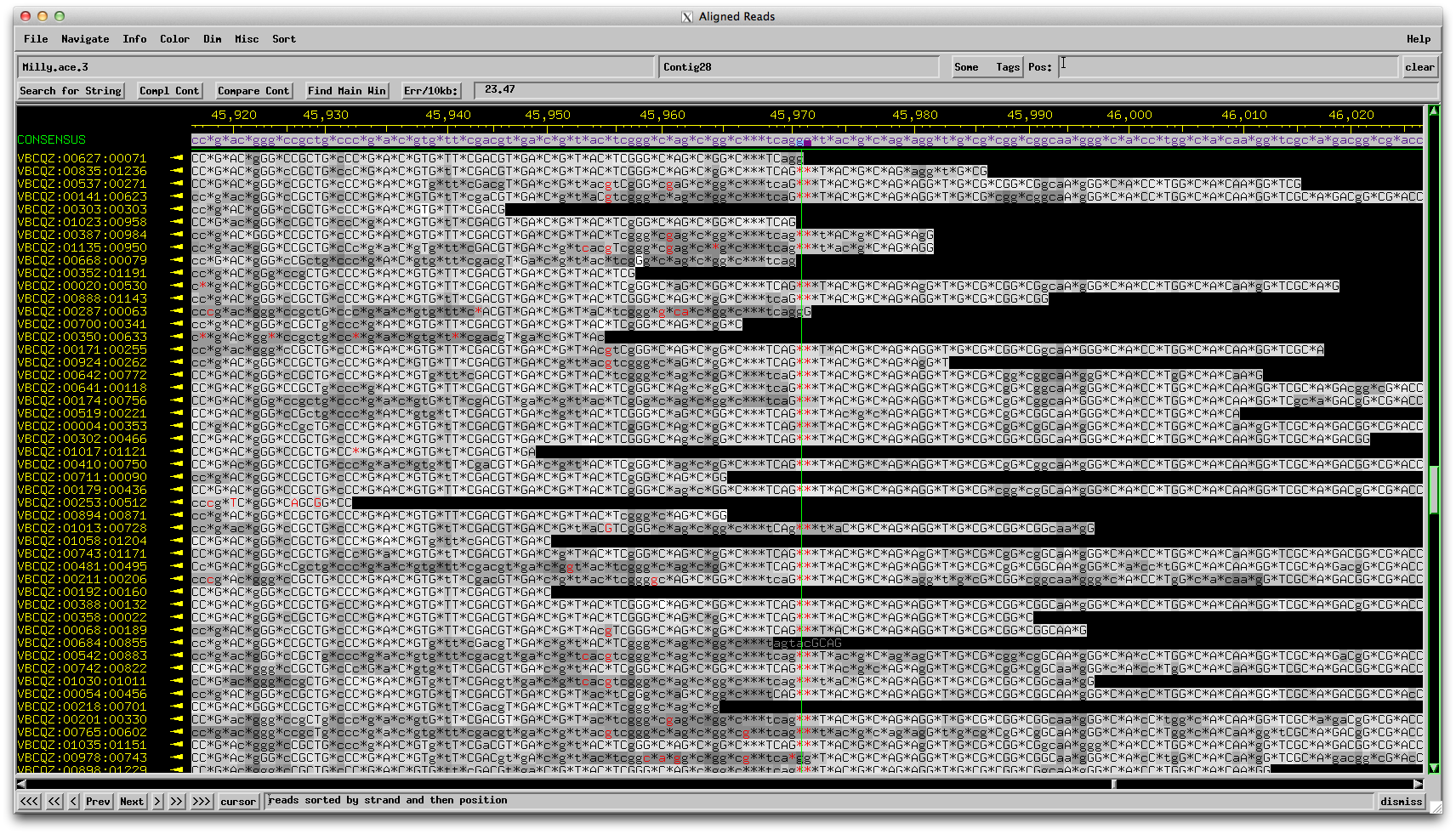 Figure 5: Milly consed assembly reverse reads: 1 G called
Figure 5: Milly consed assembly reverse reads: 1 G called
Interestingly, this error “maxes out” at 4 nucleotides. If the run of bases is longer, 5 or more, then you may have low quality reads but the errors do not seem to be as strand-specific.
We’ve seen this error in almost all genomes we’ve sequenced by Ion Torrent, including those with a range of GC contents. These S-SIDEs are also present in Ion Torrent’s own sample data sets, and have been reported in independent publications, such as the one linked to below. (See Figure 4 of this paper, e.g.)
Quail et al
What’s the Correct Sequence?
In all of the S-SIDE cases we’ve seen, and subsequently Sanger-sequenced, the correct sequence has been the strand with the longer run of nucleotides. So when one strand has 3 Gs, and one has 2 Gs, the correct consensus call is 3 Gs. This is good, because it means at least when you see one of these situations, you’ll know what the correct sequence should be.
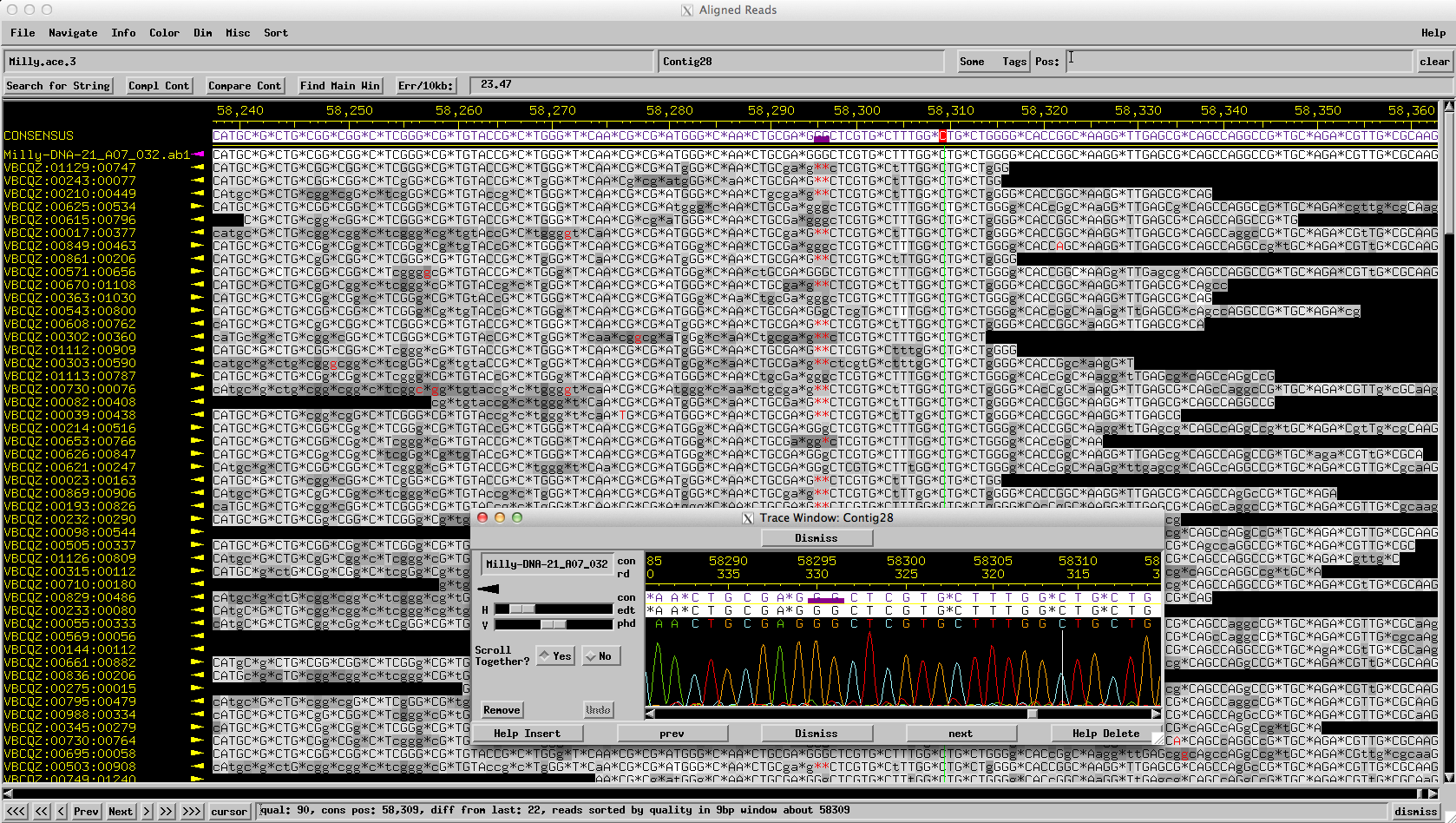 Figure 6: Milly: A Sanger read reveals the correct sequence of 3 Gs (not 1)
Figure 6: Milly: A Sanger read reveals the correct sequence of 3 Gs (not 1)
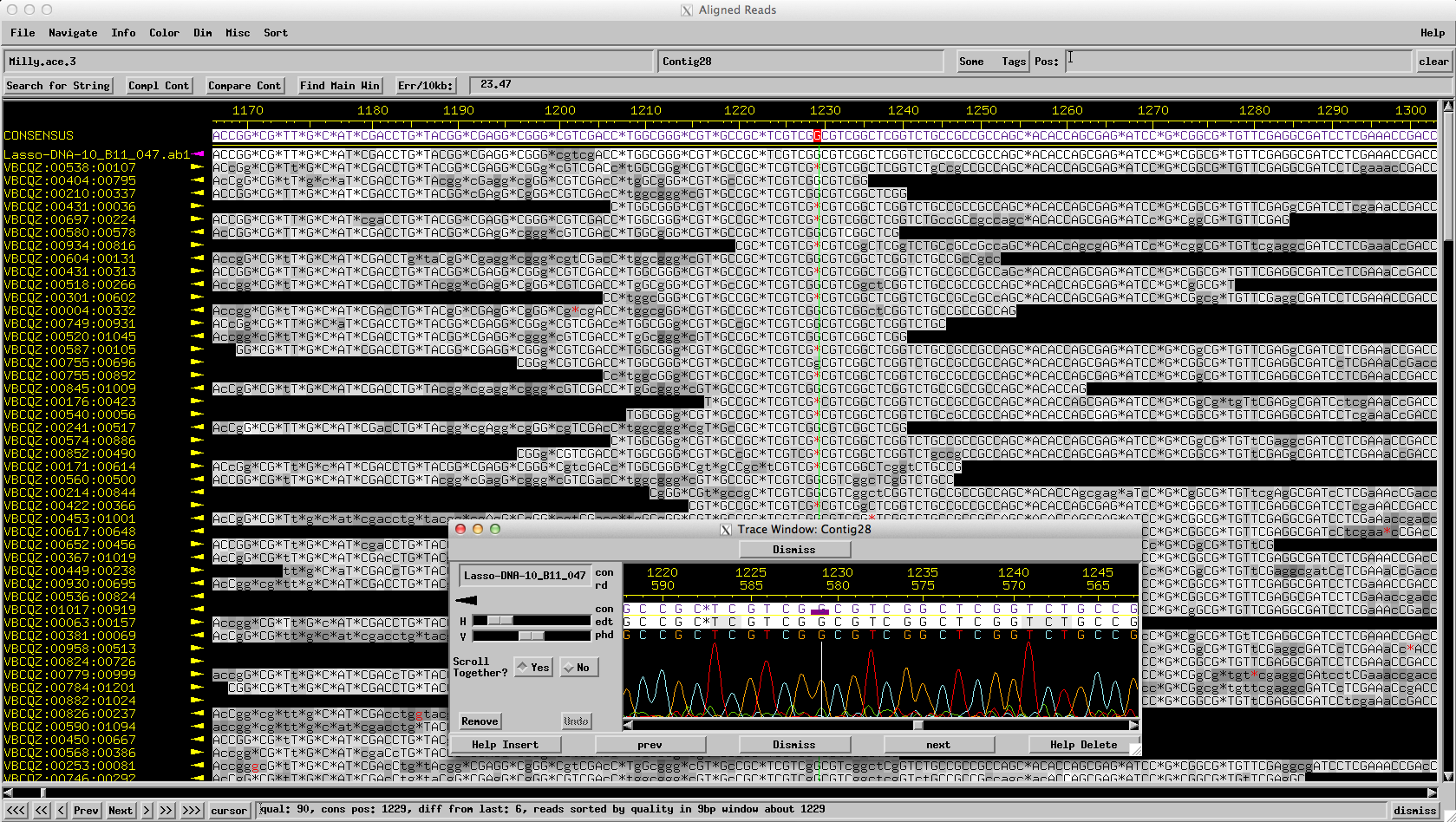 Figure 7: Lasso: A Sanger read reveals the correct sequence of 2 Gs (not 1)
Figure 7: Lasso: A Sanger read reveals the correct sequence of 2 Gs (not 1)
What’s the Problem?
During assembly, the software has to call a consensus sequence, and as you might imagine, when there’s a 50-50 conflict, it’ll sometimes choose the shorter version and sometimes choose the longer version. That means that in about half the S-SIDE cases in a given genome, the consensus will be wrong, and must be manually corrected.
Thus, every Ion Torrent PGM-sequenced genome—even if it doesn’t require any finishing bench work—will still require a fair amount of computer finishing work to correct consensus errors. As I mentioned at the outset, this is the biggest problem with sequencing genomes on the PGM.
What Can Be Done?
We use consed (which is installed on the SEA Virtual Machine) to do our finishing work here at Pitt. Charlie Bowman wrote a program called AceUtil (also installed on the 2014 SEA VM) which can flag positions in a consed assembly that have defined levels of discrepancy. You can then flip through these areas in consed and make the necessary changes to the consensus without too much trouble, though it is still a time investment.
Ion Torrent is aware of the issue, though even with the newest reagents currently available we haven’t seen much improvement in the problem. There is hope, however, that they’re going to improve the situation, at least according to Ion Torrent:
LifeTechnologies Press Release
We still like the flexibility, inexpensiveness, and easy library preps of the PGM workflow. If this newest chemistry does substantially reduce these errors, perhaps the post-sequencing part of the process will consume as little time as the pre-sequencing parts.